
سیترولینمی، بیماری است که فرم های متعدد دارد که هر کدام از آنها درمان ها و عواقب متفاوت دارند. سیترولینمی تیپ ۱ تنها یک فرم از این بیماری است. شکل دیگر بیماری CIT تیپ ۲ (CIT II) می باشد. سیترولینمی سبب ایجاد مقدار زیادی آمونیاک در بدن می گردد. CIT یک بیماری اسید آمینه محسوب می شود زیرا افراد مبتلا به CIT ، قادر به پردازش ماده زائد آمونیاک، که از تجزیه اسیدهای آمینه حاصل می گردد، نیستند. همچنین ممکن است CIT ، به عنوان بیماری چرخه ی اوره در نظر گرفته شود. این نام برای توصیف بیماری است که سیستم بدن برای از بین بردن آمونیاک از خون، به درستی کار نکند. تشخیص و درمان زود هنگام می تواند از بسیاری از پیامدهای وخیم CIT ، جلوگیری کند.

آرژینینوسوکسینیک اسیدوری (ASA)، بیماری است که باعث ایجاد مقدار زیادی آمونیاک در بدن می گردد. ASA یک بیماری اسید آمینه محسوب می شود زیرا افراد مبتلا به ASA، قادر به پردازش ماده زائد آمونیاک، که از تجزیه اسیدهای آمینه حاصل می گردد، نیستند. همچنین ممکن است ASA، به عنوان بیماری چرخه ی اوره در نظر گرفته شود. این نام برای توصیف بیماری است که سیستم بدن برای از بین بردن آمونیاک از خون، به درستی کار نکند. تشخیص و درمان زود هنگام می تواند از بسیاری از پیامدهای وخیم ASA، جلوگیری کند.

بیماری ادرار شربت افرد (MSUD)، یک بیماری است که بدن قادر به تجزیه پروتئین های خاصی نیست. این بیماری به دلیل بوی شیرین ادرار نوزادان درمان نشده، به این نام، نامگذاری شده است. MSUD به عنوان یک اختلال متابولیسم اسید آمینه در نظر گرفته شده است، زیرا افرادی که MSUD دارند، مشکل در تجزیه بعضی از اسیدهای آمینه، واحد های ساختمانی پروتئین، دارند. تشخیص زود هنگام MSUD و شروع درمان می تواند اغلب از عواقب وخیم بیماری جلوگیری کند.
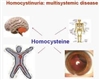
هموسیستینوری (HCY)، بیماری است که بدن قادر به تجزیه پروتئین های خاصی نیست. HCY به عنوان یک اختلال متابولیسم اسید آمینه در نظر گرفته می شود زیرا افراد مبتلا به هموسیستینوری نمی توانند برخی اسیدهای آمینه، مولکول های کوچک که پروتئین را تشکیل می دهند، پردازش کنند. این اسیدهای آمینه در بدن تجمع می یابد و می توانند مشکلات جدی برای سلامتی ایجاد کند. با این حال، اگر هموسیستینوری زود هنگام تشخیص داده شود و درمان آغاز گردد، کودکان مبتلا به هموسیستینوری، اغلب می توانند زندگی سالم داشته باشند.

تیروزینمی تیپ 1 (TYR I)، یک بیماری ارثی است که بدن نمیتواند یکی از اسیدهای آمینه موجود در پروتئین را تجزیه کند. TYR I به عنوان یک اختلال اسید آمینه در نظر گرفته می شود، زیرا افراد مبتلا به TYR I نمی توانند اسید آمینه تیروزین را تجزیه کنند. تشخیص زود هنگام و شروع درمان TYR I، اغلب می تواند از عواقب وخیم آن جلوگیری کند.
تیروزینمی یک بیماری با فرم های متعدد است، که هر کدام دارای پیامدها و درمان های مختلف است. تیروزینمی تیپ 1 (TYR I) تنها یک شکل بیماری است. فرم های مختلف بیماری عبارتند از تیروزینمی تیپ 2 (TYR II) و تیروزینمی تیپ 3 (TYR III)
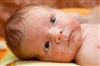
فنیل کتونوری کلاسیک (PKU)، بیماری است که بدن نمیتواند یکی از اسیدهای آمینه موجود در پروتئین را تجزیه کند. PKU به عنوان یک اختلال اسید آمینه در نظر گرفته می شود، زیرا افراد مبتلا به PKU، نمی توانند اسید آمینه فنیل آلانین را تجزیه کنند. اگر PKU درمان نشود، باعث آسیب مغزی یا حتی مرگ می شود. با این حال، اگر بیماری زود تشخیص داده شود و درمان آغاز گردد، افراد مبتلا به PKU، می توانند زندگی سالم داشته باشند.
فنیل کتونوری فرم های متعدد دارد که هر کدام از آنها درمان و پیامدهای متفاوت دارد. فنیل کتونوری کلاسیک تنها یک شکل از این بیماری است.
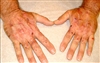
پورفیری اختلالی ارثی یا اکتسابی در ساخت هم است. هر کدام از این اختلالات الگوی خاصی از تولید بیش از حد، تجمع و دفع مواد واسطهای ساخت هم را دارند .تظاهرات بیماری بهصورت اختلال عملکرد متناوب دستگاه عصبی و یا حساسیت پوست به نور خورشید است.

Trimethylaminuria اختلالی است که در آن بدن بویی مشابه ماهی فاسد به خود می گیرد. این بیماری با نام سندرم بوی ماهی نیز شناخته می شود که به دلیل وجود آمین در تری متیل آمین (TMA) است.

پرولین یک اسید آمینه غیر ضروری برای کودکان و بزرگسالان است و می تواند از گلوتامات یا اورنیتین سنتز شود، اما در ساخت پروتئین از پرولین سنتز شده از این مسیر استفاده نمی شود زیرا پرولین بصورت روتین در رژیم ها ی غذایی وجود دارد. مقدار کمی از پرولین (51–271 μM) بصورت طبیعی در پلاسما وجود دارد. نقص های ژنتیکی در مسیر تجزیه پرولین می تواند باعث افزایش سطح پرولین(>500 μM) در پلاسما شود که این وضعیت به عنوان هیپر پرولینمیا یا پرولینوری نیز نامیده می شود که در آن اسید آمینه پرولین به درستی توسط آنزیم پرولین اکسیداز یا دلتا1_ پرولین_5_کربوکسیلات دی هیدروژناز تجزیه نشده و باعث انباشت پرولین در بدن می شود.

بیماری هارت ناپ Hartnup Disease که با نام درماتوز شبه پلاگر و همچنین ناهنجاری هارتناپ نیز شناخته میشود، جزء ناهنجاریهای متابولیسم اسیدهای آمینه است. بیماری هارتنپ که به عنوان اختلال نیزشناخته می شود، یک اختلال متابولیکی ارثی است که در آن جذب آمینو اسید های غیر قطبی به ویژه تریپتوفان که به نوبه ی خود می تواند به سروتونین و یا نیاسین تبدیل شود اختلال ایجاد می شود، در نتیجه کمبود تریپتوفان منجر به کمبود اسید نیکوتینیک و سروتونین میشود.
در بیماری های ذخیره ای گلیکوژن؛ نقص در متابولیسم گلیکوژن در بدن وجود دارد که گلیکوژن نمی تواند ذخیره شود یا فرم ذخیره نمی تواند در صورت لزوم تبدیل به گلوکز شود. گلیکوژن شکل ذخیره ای گلوکز است و فراوانترین محلی که یافت می شود کبد است که گلوکز خون را تنظیم می کند و همچنین عضلات که سبب تسهیل کار بی هوازی می شود . این گروه از اختلالات جزء تشخیصهای افتراقی هیپوگلیسمی و هپاتومگالی قرار می گیرند.

بیماری آلکاپتونوریا یک اختلال نادر متابولیکی است که به صورت اتوزومی مغلوب به ارث میرسد و در اثر نارسایی در فرایند تجزیهی اسیدآمینههای فنیل آلانین و تیروزین (اسید آمینه های آروماتیک) به وجود میآید.
مشکلات مادرزادی متابولیک آمینواسید میتواند در هر برهه ی زمانی از زندگی فرد رخ بدهد، ولی اکثرا در دوران نوزادی و در دوران آغازین کودکی ظاهر می شود. بیماران مبتلا ممکن است اختلالات رشد، علائم عصبی، مشکلات هضم، عقب افتادگی روانی-حرکتی، و طیف گسترده ای از یافته های آزمایشگاهی را از خود نشان دهند. اگر این اختلالات سریعا تشخیص داده نشده و کاملا درمان نگردند، ممکن است منجر به رشد ضعیف، عقب ماندگی ذهنی و مرگ گردند.

حداقل سه اختلال ارثی متفاوت وجود دارد که منجر به افزایش سطح اسید اوریک و بیماری نقرس(gout) می شود: نقص گلوکز-6 فسفاتاز، نقص جزئی و شدید در آنزیم هیپوگزانتین-گوانین فسفو ریبوزیل ترانسفراز و افزایش فعالیت آنزیم فسفوریبزویل پیروفسفات سنتتاز.

سطوح افزایش یافته تیروزین در سه اختلال ارثی متابولیسم تیروزین دیده می شود.تیروزینمیا نوع یک که در سال 1957 شرح داده شد ناشی از نقص در آنزیم فوماریل-استواستات هیدرولاز است. غالب افراد مبتلا کانادایی های فرانسوی زبان یا اهالی اسکاندیناوی هستند، اگر چه بیماری در گروه های قومی دیگر نیز تشخیص داده شده است. به طور کلی تخمین زده می شود که از هر 100000 نفر نوزاد زنده به دنیا آمده در کمتر از یک مورد و به طور معمول در کانادایی های فرانسوی یک نفر از هر 12500 تولد، این اختلال دیده می شود.
اریترون یک آزمایشگاه تخصصی است که از راه های مختلف میتوانید با آن در تماس باشید و پرسش ها و مشکلات خود را به آسانی با متخصصین ما در میان بگذارید.
آدرس: اصفهان، خیابان شیخ صدوق شمالی، خیابان شیخ مفید غربی
شماره تماس: 7-36631906- 031 2 -36633621 - 031
031-37134
شماره فکس: 89784728- 021 کد پستی : 76351-81647
ایمیل: med@Erythron-lab.com
شماره واتس آپ برای دریافت جواب آزمایش :
09138183947
برای هماهنگی جهت نمونه گیری در محل مورد نظر خود با شماره های زیر در ساعت مشخص تماس حاصل فرمایید:
آقای مهندس عزیزی 09131689270
تمام حقوق مطالب و محتویات این سایت متعلق به آزمایشگاه اریترون می باشد.